Are SNRIs More Effective than SSRIs? A Review of the Current State of the Controversy
Michael E. Thase, MD
Abstract and Introduction
Abstract
The selective serotonin reuptake inhibitors (SSRI) are widely considered to be the first choice for antidepressant therapy. There is evidence from inpatient studies dating to 1986, however, suggesting that the tricyclic antidepressant clomipramine, which inhibits reuptake of both serotonin and norepinephrine, may have greater efficacy than some SSRIs for severe depression. There is controversy whether the newer, better tolerated, and safer serotonin norepinephrine reuptake inhibitors (SNRIs; venlafaxine, duloxetine, and—in some countries—milnacipran and desvenlafaxine) are more efficacious than SSRIs. In addressing this controversy, this article first focuses on the limitations of randomized controlled trials (RCTs), including the factors that limit their sensitivity to detect differences between active antidepressants, and meta-analysis to examine results of large sets of RCTs. Next, the results of RCTs and meta-analyses are reviewed. Although few individual studies report significant differences, meta-analyses consistently suggest that venlafaxine may have greater efficacy than the SSRIs as a class. The magnitude of this advantage is modest (i.e., differences in remission rates of 5-10%) and no advantage has been demonstrated versus escitalopram. The advantage for duloxetine versus selected SSRIs is limited to patients with more severe depression and the RCTs are flawed by use of minimum therapeutic doses of SSRIs. No evidence of an advantage is found in RCTs of milnacipran versus SSRIs. Even a modest difference in antidepressant efficacy—if sustained—may have important public health implications for the common, disabling condition of depression. Nevertheless, differences in tolerability and cost also must be considered when choosing therapies.
Introduction
Within 5 years of the introduction of fluoxetine in late 1987, the selective serotonin reuptake inhibitor (SSRI) class of antidepressants had supplanted the tricyclic antidepressants (TCA) as the first-line pharmacotherapy for major depressive disorder throughout much of the industrialized world. There is no doubt that pharmaceutical marketing played a large role in the rapid ascendance of the SSRI class (i.e., the TCAs were no longer patented drugs and, as such, were not marketed, whereas the SSRIs were vigorously promoted). Nevertheless, the SSRIs had a number of real advantages over the TCAs, including being easier to prescribe, better tolerated, and much safer in overdose.[1] Aside from cost, which is no longer an issue with the availability of generic formulations for most of the SSRIs, another potential advantage for the TCAs was greater efficacy in severe depression (see, for example, the early review by Potter et al.).[2] However, even these data were inconsistent, with the only clear differences emerging from studies of hospitalized patients utilizing tertiary amine TCAs such as amitriptyline and clomipramine.[3,4] In retrospect, given the high prevalence of untreated depression in the early 1990s and the real limitations of the TCAs (i.e., common side effects and potential lethality in overdose), the remarkable commercial success of the SSRIs is not so surprising.
Other medications introduced in the 1980s and 1990s offered selected advantages in comparison to the SSRIs. For example, bupropion, mirtazapine, nefazodone, and—outside of the United States—moclobemide were associated with lower rates of sexual dysfunction, with mirtazapine and nefazodone also offering more rapid relief of insomnia (see, for example, Ref. 1). Among these medications, however, only bupropion has gained widespread use in the United States and that, in part, reflects the fact that it is commonly used as an adjunct or add-on therapy with SSRIs. Moreover, there was less evidence to suggest that any of these medications were more effective for treatment of depressed outpatients.
An alternate strategy for drug development focused on the so-called dual reuptake inhibitor hypothesis as a means to improve upon the efficacy profile of the SSRIs. Interest in drugs that inhibited reuptake of both serotonin and norepinephrine was fueled in part by the findings of the inpatient studies conducted by the Danish University Antidepressant Group (DUAG), in which clomipramine-the only TCA that is a strong inhibitor of serotonin reuptake—was significantly more effective than both citalopram[5] and paroxetine.[6] There was also evidence from both animal[7] and human[8] studies to suggest that combining fluoxetine and desipramine (a TCA that has strong effect on norepinephrine reuptake) could yield additive effects at both neurochemical and clinical levels. Several pharmaceutical companies therefore focused on development of selective serotonin norepinephrine reuptake inhibitors (SNRIs), with the aim of introducing antidepressants that conveyed the efficacy advantage of clomipramine while offering a side effect profile more comparable to an SSRI.
The first SNRI, an immediate release (IR) formulation of venlafaxine, was introduced in the United States in 1994, and the SNRI class now includes a more widely used extended release (ER) formulation of venlafaxine and the recently introduced metabolite drug, desvenlafaxine succinate, as well as two structurally unrelated compounds: milnacipran and duloxetine. Although milnacipran is not available in the United States, it is one of the leading antidepressants in Japan and is available in a number of European countries. This article will examine the evidence from comparative studies testing the hypothesis that SNRI therapy has greater antidepressant efficacy than SSRI, as represented by higher response and remission rates. The article has two sections: the first summarizes the methodological and statistical issues that impact on the assessment of antidepressant effects and the second reviews the evidence for—and against—the hypothesis that SNRIs are more effective than SSRIs.
Problems With Assessing Antidepressant Efficacy
Background
The randomized controlled trial (RCT) has been the "gold standard" for the evaluation of antidepressant medications for decades.[9-14] RCTs hold this exalted position because they are the best means to conduct unbiased assessments of treatment effects. One of the defining characteristics of a RCT is random assignment, which helps to ensure that treatment groups are comparable and that study participants are not "hand-picked" to match the spectrum of efficacy of one particular treatment. The other defining characteristic is experimental "control," which refers to the a priori specification of a study protocol, including inclusion and exclusion criteria, the duration of the trial, doses and titration schedule of study medications, use of standardized assessments, definition of what constitutes a therapeutic success, and a statistical analysis plan. These qualities help to ensure that the experiment is replicable. Two other features—double-blind administration of study medication and inclusion of a placebo control group—add further rigor to the experimental method, both by minimizing the impact of the expectations of the treating clinicians and participants and by providing an estimate of the likelihood that participants will improvement without a specific form of treatment.
Regulatory Approval
The pharmaceutical industry is the principal sponsor of comparative RCTs of antidepressant medications.[15] The majority of these trials are conducted as part of the registration and approval process for novel medications. These RCTs follow a sequenced paradigm that has evolved over the past 40 years to comply with the guidelines of regulatory agencies such as the United States Food and Drug Administration (FDA). A first phase of research, which is typically initiated with studies of healthy volunteers, is intended to establish the pharmacokinetic profile, maximum tolerable dose, and safety of a novel medication. Once these parameters are established by phase 1 studies, a second phase consisting of relatively small placebo-controlled trials is undertaken to determine if a medication has significant therapeutic effects. If there is evidence of efficacy in phase 2, then a new set of phase 3 RCTs are conducted to confirm efficacy versus placebo and, usually, existing standard therapies.
The FDA requires that a novel antidepressant have sufficient evidence of efficacy (i.e., drug > placebo) from at least two pivotal RCTs before that medication can be approved for general use. Although some ethicists have challenged the necessity of placebo-controlled groups for the studies of antidepressants (e.g.,[16]) the dominant view in the field is that they remain the most efficient means to establish the efficacy and safety of a novel antidepressant, and that the risks of withholding an active treatment can be minimized with careful clinical management.[17,18]
Determining efficacy versus placebo is not enough, however, and clinicians need to know as quickly as possible how a novel medication "stacks up" against standard antidepressants. Just as the TCAs were the most widely used standard of comparison for RCTs of drugs introduced in the 1980s and early 1990s, the SSRIs have now become the standard of comparison for novel antidepressants. Thus, although more costly and more difficult to conduct, the three-"arm" comparator-controlled study design provides essential information about the relative efficacy and tolerability of the novel medication. Inclusion of an active comparator in the design of a phase 3 RCT also conveys some protection for the sponsor in terms of interpretation of the results; this protection is referred to as assay sensitivity. Specifically, unless the standard drug is shown to be effective, a study in which the efficacy of the novel compound is not significantly more effective than placebo is interpreted to be "failed" rather than "negative." Thus, for the manufacturer of an investigational antidepressant, the most unwelcome outcome of a RCT is to document that the standard drug is effective but that the novel medication is not. To minimize this possibility, sponsors often utilize the minimum effective dose of the active comparator. Although this may be a sound business practice, it lessens the clinical value of the phase 3 head-to-head comparisons.
Following regulatory approval, an additional wave of RCTs is sometimes completed, which focuses comparisons versus marketplace competitors. If a novel drug is perceived to have a particular advantage, such as a more favorable effect on sleep or sexual function, such studies may be focused on subpopulations with these particular problems. Post-marketing studies often do not include placebo-controlled groups, both for ethical reasons (i.e., a placebo is more difficult to justify when both of the study medications are known to be effective) and because the aims of the study do not warrant the greater cost and complexity of a three-arm study design. In an ideal world, all phase 4 studies would be designed to provide conclusive and unbiased tests of the relative merits of the new medication versus various market leaders. More often than not, however, these studies fall short of this standard for reasons that are discussed in some detail in the next subsection.
If there is evidence of an efficacy advantage in phase 3 and postmarketing RCTs sponsored by the drug's manufacturer, a final, independent test of effectiveness in "real world" settings ideally would be conducted. Such a large (i.e., sample size > 1,000) effectiveness study would use random assignment and standardized assessment measures and dosing protocols, but would employ relatively few inclusion/exclusion criteria and would likely use "open-label"medication administration.To mitigate concerns of commercial bias, such a study could be conducted by an agency such as the National Institute of Mental Health. Although some consider large pragmatic RCTs to be at the pinnacle of the pyramid of empirical support,[14] few such studies of antidepressants have ever been undertaken and—at least to date—none have been sponsored by independent agencies.
Statistical and Methodological Issues
Statistical power refers to the likelihood that the RCT will reject the null hypothesis, assuming that there is a "true" difference between the treatments. If all other things are equal, the larger the sample size, the greater is the statistical power. Although one might wish to have 99.9% power, this lofty goal is usually not practicable (i.e., too costly). By convention, RCTs are usually planned to have at least 80% power. It is easy to determine the sample size necessary to have adequate power; software programs are now widely available for most statistical tests (for example: http://www.dssresearch.com/toolkit/spcalc/power_p2.asp ). However, such simplicity can be misleading, i.e., power calculations are highly dependent on accuracy of the investigator's assumptions and, if one or more of the assumptions are incorrect, then the chances of a failed experiment may skyrocket. For example, even the most elegantly designed study will fail if diagnoses or outcome assessments are not performed reliably. It is fortunate that a wide range of mental health professionals can learn to administer standard symptom severity measures such as the Hamilton Rating Scale for Depression (HAMD)[19] and the Montgomery Asberg Depression Rating Scale (MADRS)[20] with very high reliability. On the other hand, reliability cannot be taken for granted and inadequate attention to achieving and maintaining reliable assessments is all too often the case in large multicenter trials.[21] As Müller and Szegedi[22] have illustrated, decrements in the reliability of measurement result in progressive reductions in statistical power.
The term "effect size" refers to a standardized measure of the difference between two treatments. For a continuous variable such as the HAMD, the effect size (also known as d)[23] is calculated as the betweengroup difference divided by the pooled standard deviation. For categorical outcomes, differences in response or remission rates can be standardized in relative terms as an Odds Ratio (OR) or in absolute terms using the Number Needed to Treat (NNT) statistics (see [24]). The latter term describes the number of patients that would need to be treated with the more effective intervention in order to observe one additional response or remission. An absolute difference of 10% yields an NNT of 10, a 50% difference yields an NNT of 2. For antidepressant effects, it has been suggested that a NNT of 10 can be used as the boundary of clinical significance.[25]
It is also necessary to specify the acceptable risk that the study will yield false positive and false negative results in order to calculate power; these risks are called type I and type II errors, respectively. The type I error rate, also called alpha, is usually set at 5%, which is the origin of the hallowed P ≤ .05 convention. Type II error, also known as beta, is usually set at 20%. Thus, for a specified effect size and α-value, statistical power equals 1-ß.
It is now clear that modern RCTs of antidepressants are almost always "underpowered" because of overestimation of the expected effect size.[13] Thus, the risk of false negative findings—type II error—is much higher than previously appreciated. One reason for this problem is that placebo response rates have grown slowly over the past 30 years, without a corresponding increase in response rates to the active medications.[26] Several factors account for the greater placebo responsivity of the participants of contemporary studies, including higher expectations for benefit and differences in recruitment of study samples. Perhaps, the most important factor is almost the exclusive reliance on media advertisements, which solicit symptomatic volunteers as participants, rather than enrolling patients who have sought treatment for depression. Unfortunately, recruitment of treatment naïve symptomatic volunteers is almost a necessity for phase 2 and phase 3 studies, because few people with recurrent major depressive disorder who have previously received an effective therapy are likely to volunteer to participate in a study in which they may receive either an unproven medication or a placebo.
To make this point anecdotally, I often ask professional audiences to respond to the following question: "How many of you would enroll a depressed family member into a placebo-controlled study of a novel antidepressant?" Rarely do more than 5% raise their hands. Likewise, seldom do more than 10% raise their hands when the question is rephrased "How many of you would enroll one of your depressed patients in such a study?" If we will not enroll our patients in RCTs, who will?
The net result of these factors is that average drug versus placebo differences in contemporary antidepressant RCTs are only about one-half the size that was once expected (e.g., 10-15% differences rather than 20-30% differences).[13] A two-arm study with 100 patients per arm would have only about 50% power to detect a difference of this smaller magnitude, instead of the desired 80% (see Figure 1). Thus, it is no surprise that about one-half of the RCTs of antidepressants conducted in the 1990s failed to detect significant drug versus placebo differences.[13,27,28] When expected effect sizes are relatively small, very large studies must be undertaken to have 80% power (see Figure 1). Not only does a study with 600 patients cost three times more than a 200 patient study, but-in order to complete the research in the same time frame-it also necessitates using three times the number of clinical sites to enroll the sample. As slow enrollment is almost always an anathema to study sponsors (i.e., the "patent exclusivity clock" is ticking), industry-sponsored RCTs now routinely use a very large number of clinical sites. The necessity to enroll at 30 or even 60 sites greatly increases the complexity of a study and amplifies potential problems of reliability of measurement, which in turn may fully negate the hoped-for increase in statistical power derived from increasing the sample size.[29] In short, conducting very large studies is not a satisfactory solution to the problem of diminishing effect sizes.
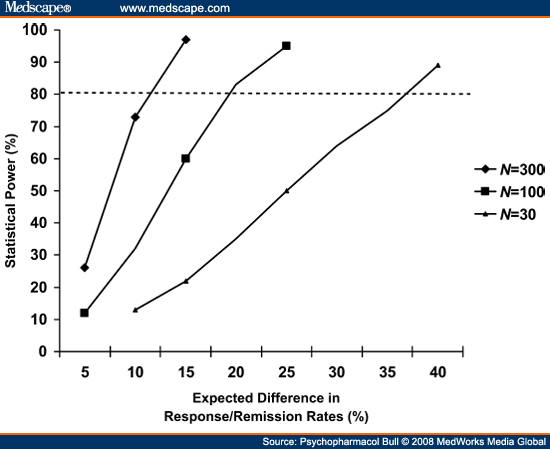 |
Figure 1. (click image to zoom) The impact of sample size and expected effect size on statistical power. Power is computed for a two-group two-tailed
χ2 test, with α = 0.05 and ß= 0.80.
|
The field is still grappling with how to best solve the problems of simultaneously delivering quantity and quality in RCTs of antidepressants. In the interim, most pharmaceutical companies have adopted a compromise: they have accepted that if one-half of their studies will fail and it is futile to try to conduct large, adequately powered studies, they will instead conduct a large number of medium-sized studies. Computation of the conditional probabilities reveals that, if each study has a 50% chance of success, a research program consisting of eight medium-sized RCTs will have about a 90% chance of success (i.e., at least two positive pivotal studies). This approach is not only wasteful and inefficient, it also is not foolproof: at least three effective antidepressants (fluvoxamine, moclobemide, and reboxetine) are not approved for treatment of depression in the United States because the manufacturer was unable to deliver the requisite two positive studies.
Implications for Comparative Studies
Although the loss of assay sensitivity has made detection of drug versus placebo differences difficult, it has even more critical implications for comparisons of the relative efficacy of different antidepressants. This is because it is virtually certain that any difference between two effective medications will be smaller than the advantage of the more effective drug versus placebo.[13] Again, the root of the problem is inadequate statistical power: if a study with 100 subjects per arm has about 60% power to detect a 15% difference between a standard antidepressant and a placebo, it will have only about 20% power to detect a difference of one-half of that magnitude between the medication and another antidepressant (see Figure 1). If the expected difference between two antidepressants is only 5%, a two-arm RCT would need to enroll more than 2,700 subjects in order to have 80% power (see Figure 2). No RCTs of this magnitude have ever been undertaken. Succinctly stated, in an era in which 50% of RCTs fail to detect a significant difference between a medication with established efficacy and an inert placebo, there is little hope that a conventional RCT will have the power to detect the difference between two active medications, even if a difference actually exists.
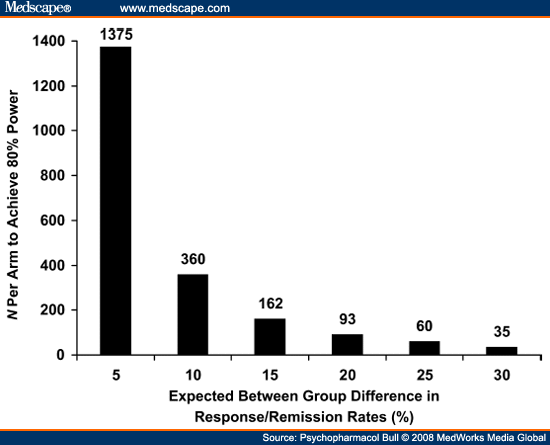 |
Figure 2. (click image to zoom) The impact of themagnitude of between-group differences on the size of cell arms needed to have 80% statistical power.
Power is computed for a two-group two-tailed χ2 test, with α = 0.05 and ß= 0.80.
|
The Role of Meta-analysis
The relatively small specific effect sizes and the absence of adequately powered head-to-head comparative studies compel researchers to consider alternate methods of gauging efficacy. The most widely used alternate approach is meta-analysis, which refers to a quantitative approach to analyze the results of a group of related studies (see [14]). The basic rationale behind meta-analysis is that a study result is simply one observation, drawn from a virtual universe of hypothetical study results, and that a more accurate estimate of the real difference between two treatments would be obtained by taking the average of a large number of studies. A corollary to this proposition is that the more comparable the studies, the more precise will be the estimate of the "true" effect size. Meta-analyses thus aim to synthesize the results of all of pertinent RCTs. In terms of modern evidence-based medicine, some consider meta-analytic confirmation of results from a series of RCTs to be necessary before a treatment warrants the highest ranking of empirical support.[14]
There are two broad types of meta-analyses. One uses the study as the unit of observation: if there are 10 relevant studies, there are 10 pairs of observations upon which to calculate the average effect size across studies. The other uses the individual patient data from a set of relevant studies to calculate the average effect of each patient (i.e., those same 10 studies might yield 1,200 patients treated with drug A and 1,200 patients treated with drug B). Although the two methods should yield the same results if performed correctly, the latter method would be preferred under ideal circumstances because it retains more information.[13] Specifically, if the data from all participants are available, the metaanalysis can address particular questions that would not be answerable from the study summary statistics, such as how do the two treatments compare in particular subsets of patients (e.g., men and women, different age groups, or different levels of pretreatment severity) or according to different definitions of outcome (e.g., remission rates instead of response rates or at different durations of therapy). However, unless the same research group has conducted all of the studies, it is difficult to obtain the individual patient data for an entire set of studies. Thus, meta-analyses utilizing study summary statistics are more usually more feasible than those utilizing the data of individual patients. Differences in study methods can also compromise the validity of pooling the results across studies. Examples include whether or not to include different end points (i.e., 6 weeks vs. 12 weeks), studies with and without placebo control, or studies of late-life depression with studies of younger adults.
Meta-analysis is not without controversy and-like the investigators of RCTs—researchers who perform meta-analysis are subject to expectancy and interpretative biases.[14] Most importantly, the validity of the results of a meta-analysis is dependent on the methods used to select, exclude, and group together particular studies. In one influential critique, Klein[30] took to task meta-analyses comparing psychotherapy and pharmacotherapy as treatments of depression, particularly those that compared the effect size of psychotherapy versus waiting list control conditions with the effect size of antidepressants versus placebo. The bottom line here is that these comparison groups are not interchangeable (i.e., a waiting list is a less powerful control than a doubleblind placebo) and, as a result, the meta-analyses are likely to overestimate the relative effectiveness of psychotherapy. Lost within the salvo of exchanges on this topic between the proponents and detractors of meta-analysis is the simple fact that—for a problem with the public health impact of major depressive disorder—so far too few adequately powered, placebo-controlled studies comparing pharmacotherapy and psychotherapy have been conducted.
The File Drawer Effect
The file drawer effect describes the selective publication of studies with positive results (i.e., the failed and negative studies are "filed away"), which presents a particular challenge to meta-analysts.[28] The most important consequence of the file drawer effect is that reviews of the published literature tend to overestimate the effects of treatments. To illustrate the magnitude of this problem, the average effect size of antidepressant therapy (versus placebo) based on meta-analyses of the published literature (e.g.,[12,26]) is approximately twice as large as it is when the results of all of the pertinent unpublished studies are taken into account.[28,31-34] To make accurate appraisals of both absolute and relative efficacy, researchers performing meta-analysis must have access to results from all relevant RCTs.
Commercial Bias
As the pharmaceutical industry is the major sponsor of RCTs of antidepressants, it should be no surprise that they are the leading contributor to the file drawer effect. Although commercial bias is readily apparent in the marketing efforts of pharmaceutical companies, it is hard to estimate the impact on systematic appraisals of treatment efficacy. On the one hand, it would be naïve to not hold a certain measure of skepticism. On the other hand, it is just plain wrong to dismiss all industry-sponsored research: this is a type of bias that is frankly much worse than the file drawer effect. The meta-analysis of Freemantle et al.[35] found that study sponsorship had relatively small impact on RCTs results: the effect was small (d < 0.10) and did not reach statistical significance. The more recent meta-analysis of Turner et al.,[28] which focused primarily on the file drawer effect, illustrates that sponsorship bias is more likely to be reflected in published reports by over-emphasis of the results of secondary analyses, particularly when no significant difference was observed on the primary outcome measure. Importantly, the impact of study sponsorship should be more transparent in future systematic reviews because all pharmaceutical companies have agreed to establish registry web sites that report summary results for all of their sponsored trials.
Comparing the Efficacy of SSRIs and SNRIs
Venlafaxine
Background. Although there is no dispute that venlafaxine is an SNRI, there has always been some uncertainty about the dose that—for the typical patient with major depressive disorder—it actually causes a clinically significant amount of norepinephrine inhibition in the brain.[36] This is because in vitro studies consistently document that both venlafaxine and its major active metabolite desvenlafaxine are substantially weaker inhibitors of norepinephrine uptake transporters than serotonin uptake transporters (see, for example,[37]). Although there is evidence that these drugs have some measurable physiological effects indicative of norepinephrine reuptake inhibition across the full dose range (i.e., 75-375 mg/day), there continues to be a pervasive view that, at lower therapeutic doses, venlafaxine is essentially an SSRI and that upward dose titration is needed to produce sufficient norepinephrine reuptake inhibition in the brain to enhance antidepressant activity.[36,38]
Several lines of clinical evidence support this perception. First, the effects of venlafaxine on blood pressure, which are presumed to be at least partly mediated by norepinephrine reuptake inhibition, are strongly dose-dependent, and at minimum therapeutic dose (75 mg/day) the effects of venlafaxine on blood pressure are similar to those of placebo.[39,40] Second, venlafaxine is the only modern antidepressant to show a clearcut dose-response relationship in RCTs.[41-44] Many of the side effects of venlafaxine that are presumed to be mediated by norepinephrine reuptake inhibition, including dizziness, dry mouth, increased pulse, and increased sweating, show similar dose-dependence.[45] Third, several early studies of low-dose venlafaxine therapy failed to demonstrate any difference in efficacy versus comparably low doses of SSRIs.[46,47] As there is not yet a reliable method to measure norepinephrine uptake inhibition in the living brain (i.e., a reliable, nontoxic radioligand is not available) and studies utilizing peripheral or ex vivo assays of noradrenergic effects have yielded conflicting results (see[36]), this issue cannot be definitively resolved.
Evidence From RCTs. As the first member in class, venlafaxine is also most extensively studied in RCTs versus SSRIs as active comparators. Although there has been evidence of an efficacy advantage for venlafaxine in RCTs versus fluoxetine for over a decade (e.g.,[48-49]), this issue was pushed to the forefront in 2001 following the publication of a meta-analysis of individual patient data.[33] Working in collaboration with the medical director and statistician of the Wyeth research group, I "pooled" the results of the first eight double-blind RCTs contrasting venlafaxine with SSRIs in major depressive disorder; no study meeting these criteria was excluded. The meta-analysis included data from 851 patients treated with venlafaxine (dose range: 75-375 mg/day; modal dose across studies = 150 mg/day) and 748 patients treated with SSRIs (fluoxetine, n = 563, modal dose: 40 mg/day; paroxetine, n = 160, dose range: 20-40 mg/day; fluvoxamine, n = 34, dose range: 100-200 mg/day). As only four studies included placebocontrolled group, there were a smaller number of patients (n = 446) randomly assigned to placebo. The primary outcome was intent to treat remission rates at week 8 or study endpoint, as defined by a HAMD score of ≤7. Results indicated a significant advantage for venlafaxine over SSRIs (remission rates: 45% vs. 35%), with SSRIs superior to placebo by a comparable margin (remission rates: 35% vs. 25%) (see Figure 3). The report also included a number of secondary analyses that confirmed that the advantage of venlafaxine was evident across all commonly used definitions of response and remission and was not dependent on any particular study characteristic or any single study.[33] Although the absolute difference in remission rates favoring venlafaxine over the SSRIs was modest (10%), measures of relative efficacy—whether effect sizes were expressed as an OR (1.52) or NNT (10)—were large enough to be considered clinically significant.
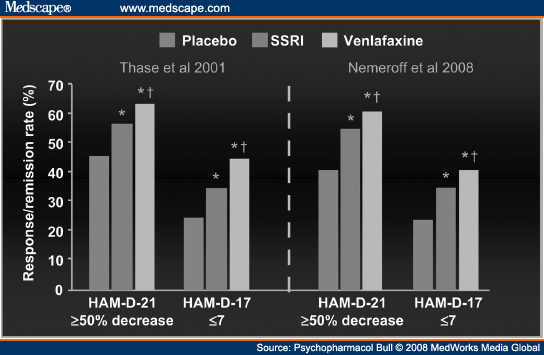 |
Figure 3. (click image to zoom) Remission rates in theoriginalmeta-analysis of individual patientdata from eightdouble-blind studies comparing
venlafaxine and various SSRIs andthe subsequent 34 study replication (source:ref. 33, andref. 63).
|
Subsequent analyses of this data set indicated that venlafaxine was also significantly more effective than SSRIs with respect to reductions in HAMD and MADRS scores[50] and in terms of fewer days spent depressed.[51] When these findings were incorporated in a cost-effectiveness analysis, the greater acquisition cost of venlafaxine XR relative to "name brand" SSRIs was more than offset by its greater efficacy.[52] The efficacy advantage of venlafaxine was also consistent across various age groups, from young adulthood to later life.[53] Of note, a subsequent analysis revealed an interesting three-way interaction between antidepressant type, age, and gender.[34] Specifically, the remission rates of patients treated with SSRIs and placebo—but not venlafaxine—were significantly lower among the subset of women aged 50 and older; this difference was particularly large among those not taking hormone replacement therapy (HRT).[34] In other words, the NNT favoring venlafaxine therapy for most patients ranged between 9 and 11, whereas the advantage for postmenopausal women not taking HRT was markedly larger with a NNT of 4.
The report of Thase et al.[33] received a fair amount of scrutiny and has drawn some criticism. Perhaps the most important limitation is that the results were heavily dependent on the venlafaxine versus fluoxetine comparisons. In fact, less than one-third of the SSRI group was treated with paroxetine or fluvoxamine and there were no patients treated with sertraline or citalopram (escitalopram was not available at that time). Although a secondary analysis did demonstrate a statistically significant advantage for venlafaxine over the pooled "nonfluoxetine" group, the remission rate of the patients treated with paroxetine or fluvoxamine was not significantly greater than that of the patients treated with placebo,[33] which means that confidence in this particular secondary comparison is limited by a lack of assay sensitivity.
A second criticism is that the meta-analysis was based on data from 6 to 8 week acute phase therapy trials. This leaves open the possibility that the patients treated with SSRIs may have "caught up" with the venlafaxine- treated group after an additional month or two of treatment. As an effective course of antidepressant therapy should be continued for months or even years, it would have been much more compelling to show that the efficacy advantage of venlafaxine is sustained and not simply a transient consequence of a 1- or 2-week acceleration in speed of response. The potential importance of this criticism is amplified by fact that the meta-analysis included a disproportionate number of patients treated with fluoxetine, which—because of the extremely long time to steady state for the active metabolite norfluoxetine—may have a somewhat slower onset of action than for the remainder of the SSRIs.[54] This point was illustrated by the results of a large double-blind study comparing venlafaxine and fluoxetine in recurrent depression, in which temporal trends favoring venlafaxine early in the acute phase protocol dissipated by week 10 and were nonexistent during the subsequent continuation phase.[55]
A third criticism is that the studies included in the meta-analysis did not systematically exclude patients with a history of nonresponse to other SSRIs. As venlafaxine was a novel compound when these studies were conducted and, in most countries, there were several other SSRIs available, the modest "across the board" advantage for venlafaxine might actually have concealed two different response patterns: therapeutic equivalence for treatment naïve patients and a larger advantage for those with a history of SSRI nonresponse (see, for example,[56]). Although such a finding would still have significant clinical implications, it would be less meaningful than a broader advantage as a first-line therapy.
Other criticisms concerned the possibility of bias in selection of studies. Warner[57] used the results of the individual studies to construct a funnel plot, which is a method to examine the observed effect sizes of a set of studies (x axis) in relation to their sample size (y axis). If a metaanalysis is appropriately inclusive, the plot should resemble an inverted funnel, with greater dispersion of results among smaller studies and, with increasing sample size, more precise estimation of the average difference. On the basis of his visual inspection of the plot, Warner[57] suggested that Thase et al.[33] omitted some unknown number of smaller studies and, hence, the results of the meta-analysis may not be representative of the broader body of evidence. Although this criticism is factually accurate, Warner seemed to overlook the fact that Thase et al.[33] restricted their meta-analysis to specific set of relatively large doubleblind RCTs that had been subjected to regulatory review and for which individual patient data were available. In other words, as clearly outlined in their report, Thase et al.[33] had indeed excluded all such smaller studies. Moreover, Warner[57] incorrectly drew the funnel plot, plotting effect sizes according to a standard (base 10) scale rather than the logarithmic scale that should be used for ORs. This error distorted the shape of the plot and exaggerated the asymmetric appearance of the funnel plot. With respect to the exclusion of studies, Thase et al.[33] did identify and tabulate the results of nine other RCTs, which were either excluded for methodologic reasons (i.e., open-label designs) or were finished after the original meta-analysis was completed. Although these studies used a variety of different definitions of remission, calculation of the betweengroup differences revealed an average effect that was slightly larger than that of the original meta-analysis.
Several other critics suggested that inclusion of data from unpublished studies weakened the validity of the findings because it was not possible to examine the degree of experimental rigor or to uncover methodological flaws.[58,59] Although it is true that meta-analysts sometimes grade studies according to methodologic rigor, all of the studies included in the Thase et al.[33] report were randomized, double-blind trials with outcome assessed according to the HAMD and, as such, would have received at least a passing grade in terms of methodologic rigor. Even more importantly, these critics did not seem to understand that it is the exclusion of unpublished (and usually failed) studies that is the basis of the "file draw" effect and that to knowingly exclude the very studies that the manufacturer of venlafaxine had chosen not to publish would have been a far greater threat to the validity of the meta-analysis than to include them.[13]
The number of RCTs comparing venlafaxine and SSRIs has continued to proliferate and, as of May 2008, there are nearly 50 comparative studies, including more than 20 studies of SSRIs other than fluoxetine. This permits the most extensive assessment of relative efficacy among all the modern antidepressants. A funnel plot of remission/response rates from all known studies circa May 2008 is provided in Figure 4. This plot illustrates that ∼80% of the head-to-head comparisons yielded numeric advantages in favor of the SNRI.With respect to the concern expressed by Warner,[57] this plot provides one of the best approximations of the desired funnel shape in the published literature. A number of updated meta-analyses examining the efficacy of venlafaxine and SSRIs have been completed, including both conventional meta-analyses of study summary results[25,61,62] and individual patient data.[63] Collectively, these meta-analyses confirm a modest, statistically significant advantage favoring venlafaxine over the SSRI comparators. Importantly, the meta-analysis of Cipriani et al.,[25] which focused specifically on comparative studies of fluoxetine, confirmed the advantage using the strict methods of the Cochran Collaboration review group.
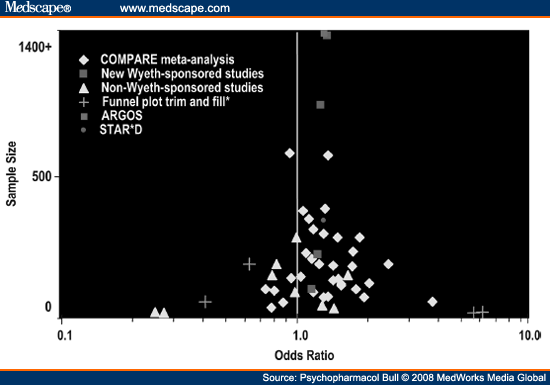 |
Figure 4. (click image to zoom) Funnel plot trim and fill analysis including all studies comparing venlafaxine and SSRIs. Markers represent point
estimates of odds ratios for remission plotted according to sample size. Overall adjusted effect odds ratio was 1.26 (95% confidence interval: 1.16–1.38). The funnel plot trim
and fillmethod identifies excess statistical outliers on either side and "fills" the opposite side with theoretical studies accordingly to balance the funnel. An adjusted weighted
effect size is then calculated from the revised plot. No evidence of statistically significant selection bias was detected with the linear regression test (Z = -.19,
P= .42) and rank correlation test (Z = .05, P = .48).[60]
|
The results of the largest of these meta-analysis, which replicated the methods of Thase et al.[33] using the individual patient data from 34 double-blind RCTs conducted by the manufacturer of venlafaxine, were recently published.[60] In the replication study, the average difference favoring venlafaxine was somewhat smaller than in the first report (across all studies, the absolute difference in response/remission rates was about 6%; OR = 1.3; NNT = 17; d = 0.20), it is also true that such effect sizes are ∼60% of the effect sizes of SSRIs versus placebo in the studies conducted by SSRI manufacturers (see, for example,[31,64-65]).
Returning to some of the other criticisms of the original meta-analysis, new data does permit the question about the relative efficacy of venlafaxine and SSRIs in treatment-resistant depression. Specifically, two RCTs employing more pragmatic designs compared venlafaxine and SSRIs following nonresponse to another standard antidepressant.[66,67] The study of Rush et al.,[67] which was a component of a larger research initiative known as Sequenced Treatment Alternative to Relieve Depression (STAR*D), included the pairwise comparison of venlafaxine (n = 250; mean dose: 194 mg/day) and sertraline (n = 238; mean dose: 136 mg/day) following prospectively observed nonresponse or intolerance to up to 12 weeks of therapy citalopram (maximum dose 60 mg/day). The study utilized random assignment to treatment and independent "blinded" clinical evaluators, although treatment was administered open label. Neither treatment was particularly effective, with remission rates of only 26% and 19% observed for the SNRI and SSRI, respectively. As the study was designed to have the statistical power to detect 15% between-group differences, this more modest advantage was not statistically significant. The second trial, which enrolled 3,097 patients recruited from clinical practices of 422 Spanish psychiatrists, utilized a simpler design, in which patients with a history of nonresponse or intolerance to "conventional antidepressants" were randomly assigned to up to 6 months therapy with either venlafaxine or the treating psychiatrist's choice of a SSRI or mirtazapine. Intent-to-treat remission rates at month 6 or study endpoint were 59% for the patients treated with venlafaxine and 52% for those receiving other antidepressants. In such a large study, this difference was significant, as were the pairwise comparisons between the venlafaxine group and the subgroups treated with fluoxetine, sertraline, paroxetine, and citalopram. When the results of these two studies are taken together, it appears that the advantage of venlafaxine observed in patients with a history on nonresponse in "real world" settings is about the same magnitude as reported in more highly controlled RCTs of unselected patients groups and, as such, it seems unlikely that inclusion of patients with a history of SSRI nonresponse biased the results of the efficacy trials.
Possible Clinical Exceptions. Two potential exceptions to the pattern of greater overall efficacy have emerged that warrant comment. The first pertains to studies of bipolar depression.[68,69] In these trials, there was no clear-cut pattern of an efficacy advantage, yet there was suggestive evidence of greater risk of treatment emergent affective switch (TEAS) despite concomitant therapy with mood stabilizers as compared to patients treated with paroxetine[68] or sertraline.[69] As the TCAs also have been found to be associated with a greater risk of TEAS than the SSRIs,[70] these findings suggest that the noradrenergic effects of venlafaxine may be problematic for at least a subset of more vulnerable bipolar patients.[45]
The second potential exception pertains to escitalopram, the last member of the SSRI to receive regulatory approval.[71] As the active enantiomer of citalopram, escitalopram is the most selective of the SSRIs, yet it appears to differ from the other members of this class because of its effects on a second allosteric mechanism that modulates drug dissociation from the serotonin transporter (see, for example, Ref. [72]). Indeed, a series of pooled analyses of individual patient data from RCTs contrasting escitalopram and citalopram found suggestive evidence of greater efficacy, particularly among more severely depressed patients treated at the higher therapeutic dose (20 mg/day) of escitalopram (reviewed by Thase).[71] A recent meta-analysis of RCTs comparing escitalopram with citalopram and other SSRIs reached a similar conclusion.[73] Across studies, the magnitude of this advantage appears to be on the order of a 5% to 10% difference in response or remission rates, i.e., an advantage comparable to that observed for venlafaxine versus the other SSRIs. These data also suggest that there may be sufficient heterogeneity within the SSRI class to call into question the validity of grouping these drugs together.
With these findings in mind, the results of the two head-to-head RCTs directly comparing venlafaxine and escitalopram are of some interest.[74,75] The findings of both studies indicated that the drugs were comparably effective, although neither study was large enough to formally test "noninferiority" (i.e., to conclude with reasonable certainty that no difference exits). As discussed previously, without adequate statistical power, the endemic problem of type 2 error cannot be discounted. However, the results of a pooled analysis of individual patient data from these two trials suggested that escitalopram might actually be more effective than venlafaxine for treatment of more severely depressed patients.[76]
Interpretation of the pooled data set is compromised by an asymmetry in dosing strategies used in one of the studies.[75] Specifically, this study utilized a rapid titration protocol in which both medications were increased to maximum approved dose within 8 days of starting study therapy. The asymmetry is that the dose of escitalopram was doubled in that time frame (i.e., increased from 10 to 20 mg/day), whereas the dose of venlafaxine ER was tripled (i.e., from 75 to 225 mg/day). As a result, there were uncharacteristically large differences in tolerability favoring escitalopram, including a fourfold difference in the proportion of patients who dropped out due to intolerable side effects (venlafaxine 16% vs. escitalopram 4%). Nevertheless, it can be concluded that—at the least—there is no evidence that venlafaxine is more effective than escitalopram.
Summary. The efficacy advantage of venlafaxine compared to the SSRIs as a class is small but replicable with the likely exception of escitalopram. If practice was based solely on this finding, the improvement in patient outcomes would be modest and result in one more case of response or remission for every 10-15 patients treated. Although Trivedi et al.[52] established that venlafaxine XR was a cost-effective option versus "name brand" SSRIs, and it is uncertain if a 5 to 10% difference in remission rates would be sufficient to offset costs for first-line use in an era in which multiple generic formulations of fluoxetine, paroxetine, citalopram, and sertraline are available. Beyond cost, additional considerations, including the risk of treatment-emergent high blood pressure and epidemiologic evidence suggestive of greater toxicity in overdose (e.g., Ref. 77), further justify the practice in some guidelines and formularies to adopt a "generic SSRI first" policy. With this restriction in mind, there is good evidence from several recent studies that venlafaxine is an effective therapy following nonresponse or intolerance to SSRIs.[66,78,79]
Desvenlafaxine
Just introduced in the United States in early 2008, desvenlafaxine succinate (DVS) is a commercially formulated version of the O-desmethylmetabolite of venlafaxine. As such, DVS is to venlafaxine as nortriptyline and desipramine are to amitriptyline and imipramine. Among individuals with normal hepatic metabolism via Cytochrome P450 2D6, the O-desmethyl metabolite accounts for approximately twothirds of the combined plasma concentration of the parent drug and the metabolite.[36] Thus, because venlafaxine and O-desmethylvenlafaxine are roughly equipotent, the O-desmethyl metabolite is likely to account for the majority of the clinical effects in patients treated with venlafaxine. The therapeutic profile of DVS is thus likely to be quite similar to the venlafaxine. The major potential advantage for DVS over the parent compound is a simpler dosing regimen (i.e., it is anticipated that most patients will be treated with either 50 or 100 mg/day); it is also possible that a somewhat greater potency for NE reuptake inhibition may result in a more potent effect at lower doses.[80] Comparative studies versus escitalopram have been undertaken, but results are not yet available.
Milnacipran
Background. Milnacipran, which is not yet available in the United States, was the second SNRI to be introduced and it has gained widespread use in Japan and several other countries. Unlike venlafaxine, milnacipran is a more potent inhibitor of norepinephrine reuptake at minimum therapeutic doses (i.e., 25 mg twice daily) and may require upward titration (i.e., up to 100 mg twice daily) in order to affect significant inhibition of serotonin reuptake in the central nervous system.[81]
Evidence From RCTs. Although there are a number of uncontrolled reports and case series in the literature that suggest that milnacipran may have an efficacy advantage compared to SSRIs,[82-84] neither the results of an early pooled analysis supported by the manufacturer[85] nor any of the six double-blind RCTs in the peer reviewed published literature reported a significant advantage in response or remission rates.[86-91] It is not known if other comparative studies have been completed but not published.
Papakostas and Fava[92] conducted a meta-analysis of the six published studies, using response rates on the MADRS as the outcome of interest. The studies included a total of 1,082 patients with major depressive disorder; treatment duration ranged between 4 and 12 weeks. The effects of milnacipran and SSRIs were almost identical, with average MADRS response rates of 58.9 and 58.3%. Response rates on the HAMD, which were reported in five of the studies, likewise revealed no difference in efficacy (milnacipran: 59.7%; SSRIs: 57.5%).
Summary. There is no evidence from RCTs that milnacipran is more effective for the average patient than the SSRIs that have been studied. The lack of studies of sertraline and escitalopram, the relatively low doses of fluoxetine and citalopram used in several of the comparative studies, and the possibility of publication bias collectively suggest that this estimate of comparable efficacy is, if anything, optimistic. If the doses of milnacipran that were utilized in these studies were sufficiently high to achieve a meaningful "dual reuptake" inhibitor effect (i.e., ≥80% occupancy of serotonin transporters in the brain), these findings cast major doubt on the hypothesis that the SNRIs—as a class—are more effective than the SSRIs.
Duloxetine
Background. Duloxetine, which was introduced in the United States in 2004, is promoted by the manufacturer as a "balanced" SNRI. This refers to the fact that in vitro studies demonstrate duloxetine is a significantly more potent norepinephrine reuptake inhibitor than venlafaxine and a significantly more potent serotonin reuptake inhibitor than milnacipran.[93-95] In practical terms, the benefit of such balance could be that duloxetine therapy would be characterized by a clinically relevant effect on both neuronal systems at the minimum therapeutic dose (i.e., 60 mg) and, as such, might require less titration to achieve optimum therapeutic effect than either milnacipran or venlafaxine.[96,97] Consistent with this notion, placebo-controlled studies of duloxetine therapy have documented a relatively flat dose-response curve from 60 to 120 mg/day.[98,99]
It is noteworthy that, despite more potent inhibition of norepinephrine uptake than venlafaxine, duloxetine therapy is not associated with increased rates of treatment-emergent high blood pressure.[100] In fact, rates of sustained elevations of diastolic blood pressure in RCTs were less than 1% greater for patients taking the maximum dose of duloxetine (i.e., 60 mg bid) than observed among patients treated with placebo.[100]
Evidence From RCTs. The research program that led to regulatory approval of duloxetine in the United States and Europe included six double- blind RCTs with SSRI comparison groups (see[96,101]). All six studies utilized fixed dose designs and were placebo-controlled. Across studies, three doses of duloxetine (20, 40, and 60 mg bid) were compared versus fluoxetine 20 mg/day (2 studies) or paroxetine 20 mg/day (4 studies). Only one of the studies reported a significant difference between duloxetine (80 mg/day arm) and paroxetine,[102] although nonsignificant trends favoring the SNRI were observed in several other RCTs.[103,104] Inspection of effect sizes (i.e., drug versus placebo differences) across studies similarly suggested a small advantage for duloxetine.[105]
The individual patient data from all six RCTs were made available for a meta-analysis.[101] To summarize, in the overall study group (n = 1833), the remission rates of patients treated with duloxetine 40-120 mg/day were 2% greater than those treated with fluoxetine or paroxetine (see Figure 5, panel a); this difference was not statistically significant. It is noteworthy that these studies utilized a relatively low inclusion severity score on the HAMD (patients with scores ≥14 were eligible), which meant that about 40% of the patients who participated in these trials were so mildly depressed that they typically have been excluded from phase 3 RCTs. For this reason, the meta-analysis included a planned comparison delimited to the subgroup of 1,044 patients who scored ≥19 on the HAMD prior to treatment. Among these moderately to severely depressed patients, the advantage of duloxetine therapy versus the SSRIs was a difference in remission rates of 7.3% (see Figure 5, panel b). This difference, which was almost identical to that reported by Thase et al.[33] in the initial report on venlafaxine, was statistically significant.
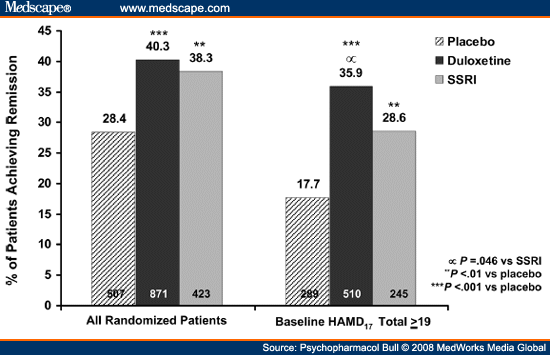 |
Figure 5. (click image to zoom) Meta-analysis of individual patient data from six double-blind studies comparing duloxetine and SSRIs. The remission
rate difference between duloxetine and the SSRIs (fluoxetine or paroxetine) was not significant in the overall population (panel a), but was among the patients with pretreatment
hamd scores of ≥19 (panel b) (source: ref. 101).
|
Three characteristics of this initial group of comparative RCTs of duloxetine warrant additional comment. First, the advantage of the SNRI among more severely depressed patients was not the result of "worse than usual" performance of the SSRI comparators. Second, all six of the duloxetine studies used four-arm fixed dose designs, which are significantly less sensitive to detecting drug-placebo differences than two- or three-arm flexible dose designs.[106] Thus, the attempts of Eckert and Lancon[107] and Vis et al.[108] to compare effect sizes of the duloxetine studies with those from a set of venlafaxine studies, which are predominately flexibly dosed, are likely to have underestimated the efficacy of duloxetine. Third, none of the early comparative trials studied the minimum therapeutic dose of duloxetine (60 mg/day), whereas all six of the trials used the minimum therapeutic dose of the SSRI comparator (i.e., 20 mg per day of fluoxetine and paroxetine). Fourth, none of the early studies utilized citalopram or escitalopram as the comparator.Additional RCTs were therefore needed to evaluate the relative efficacy of the 60 mg dose of duloxetine and to ensure that any advantage apparent for the SNRI is not a consequence of the SSRIs and doses studied.
Three studies have subsequently contrasted duloxetine 60 mg/day versus escitalopram 10 to 20 mg/day.[109-111] It is not yet possible to pool the data from these trials (two were conducted by the manufacturer of escitalopram and one by the manufacturer of duloxetine), though inspection of the results of the individual studies would indicate that escitalopram was at least as effective as duloxetine, both during the initial acute phase of therapy[109,110] and across up to 6 months of continued therapy.[111,112] Moreover, advantages favoring the SSRI were observed on some secondary outcome analyses in the two studies conducted by the manufacturer of escitalopram.[109,111] Across studies, escitalopram also showed significant tolerability advantages,[109-112] both in terms of attrition rates and the incidence of most common side effects other than the incidence of sexual side effects.[113]
Conclusions
The controversy about the relative efficacy of different types of antidepressants continues in part because there are numerous problems that limit the sensitivity of RCTs to detect efficacy differences between active antidepressants. Although there is only a partial solution to the problem of inadequately powered studies, meta-analysis permits quantitative synthesis of results from a group of relevant RCTs comparing various types of antidepressants. Meta-analyses of RCTs comparing the SSRIs with two of the SNRIs—venlafaxine and duloxetine—may have greater efficacy, though there is essentially no evidence that either of these drugs is superior to escitalopram. For venlafaxine, which is so far the most extensively studied SNRI, the magnitude of this advantage versus SSRIs other than escitalopram is modest in unselected patient groups (NNT values range between 10 and 15) and appears to be greater versus fluoxetine than other members of the class. For duloxetine, the advantage versus paroxetine and fluoxetine appears to be limited to more severely depressed patients, and confidence in this finding is limited by the fact that the studies used minimum therapeutic doses of SSRIs. With respect to the other two SNRIs, there are no data yet available to evaluate the relative efficacy of desvenlafaxine. Available data suggest that there is no efficacy advantage for milnacipran. The latter finding, coupled with the failure of both venlafaxine and duloxetine to significantly separate from escitalopram, highlights the limited clinical utility of comparisons across particular classes of medication.
Reprint Address
Michael E. Thase, University of Pennsylvania School of Medicine, 3535 Market Street, Suite 670, Philadelphia, PA
No comments:
Post a Comment